cdBEST -
Chromatin Domain Boundary Element Search Tool
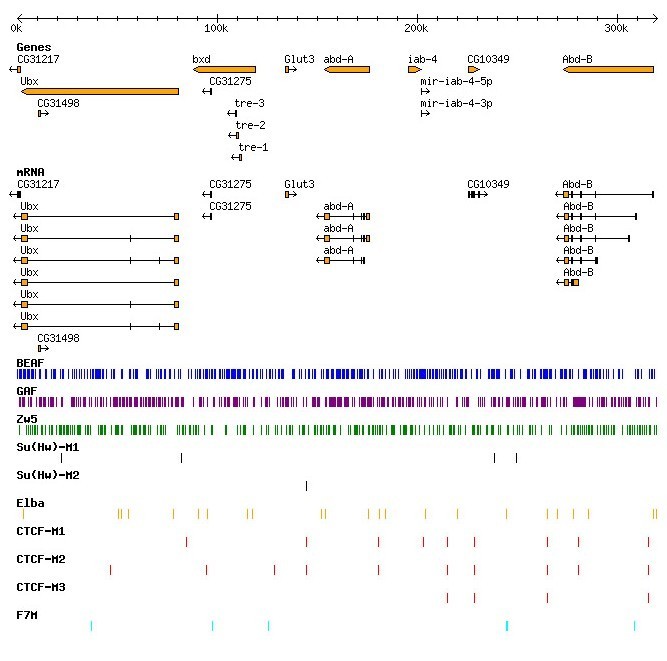
Chromatin domain boundary elements prevent inappropriate interaction between distant or closely spaced regulatory elements
and restrict enhancers and silencers to correct target promoters. In spite of having such a general role and
expected frequent occurrence genome wide, there is no DNA sequence analysis based tool to identify boundary
elements. We developed the chromatin domain Boundary Element Search Tool (cdBEST) to identify boundary elements.
cdBEST uses known recognition sequences of boundary interacting proteins and looks for ‘motif clusters’. Using
cdBEST, we identified boundary sequences across 12
Drosophila species. Of the 4576 boundary sequences identified in
Drosophila melanogaster genome, >170 sequences are repetitive in nature and have sequence homology to
transposable elements. Analysis of such sequences across 12
Drosophila genomes showed that the occurrence of repetitive sequences in the context of boundaries is
a common feature of drosophilids. Enhancer-blocking assays on a subset of the cdBEST boundaries show that 80%
of them indeed function as boundaries in vivo. cdBEST thus provides a better understanding of chromatin domain
boundaries in
Drosophila and sets the stage for comparative analysis of boundaries across closely related species.
C-State -
'C' the Chromatin State
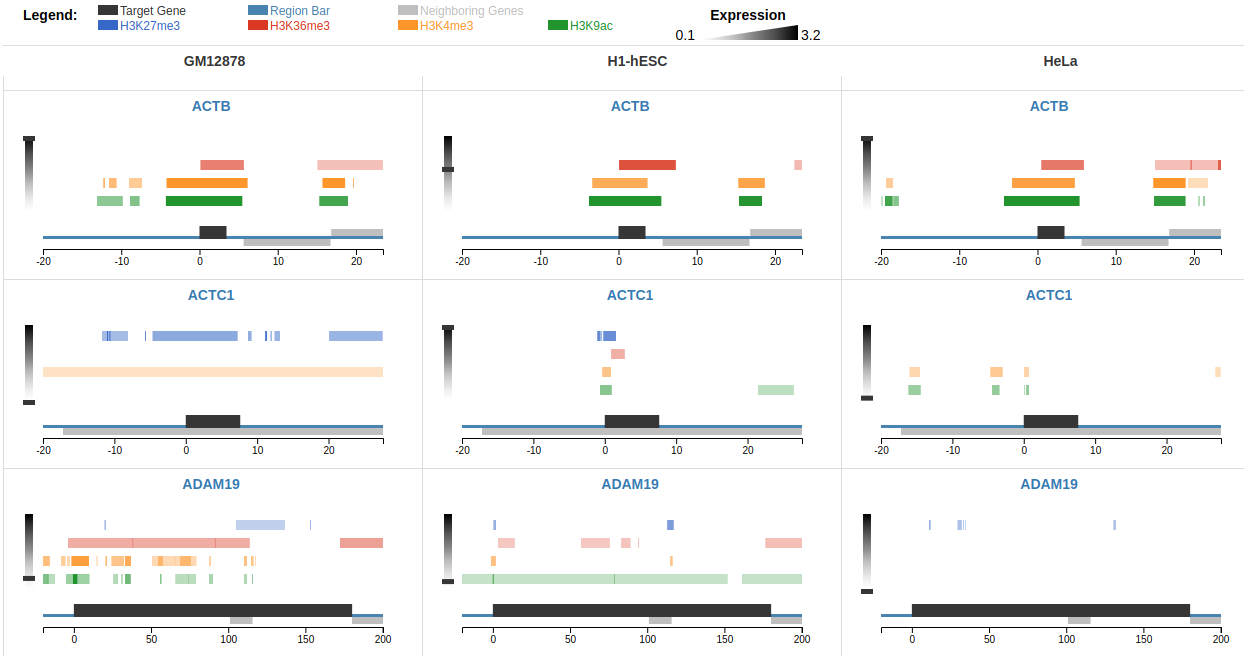
Comparative epigenomic analysis across multiple genes presents a bottleneck for bench biologists working with NGS data. Despite
the development of standardized peak analysis algorithms, the identification of novel epigenetic patterns and
their visualization across gene subsets remains a challenge. We developed a fast and interactive web app, C-State
(Chromatin-State), to query and plot chromatin landscapes across multiple loci and cell types. C-State has
an interactive, JavaScript-based graphical user interface and runs locally in modern web browsers that are
pre-installed on all computers, thus eliminating the need for cumbersome data transfer, pre-processing and
prior programming knowledge. C-State is unique in its ability to extract and analyze multi-gene epigenetic
information. It allows for powerful GUI-based pattern searching and visualization. Its potential for identifying
user-defined epigenetic trends in context of gene expression profiles is demonstrated at the
Het C-State page and in the case studies in the
user manual.
GET -
Genomes Exploration Tool
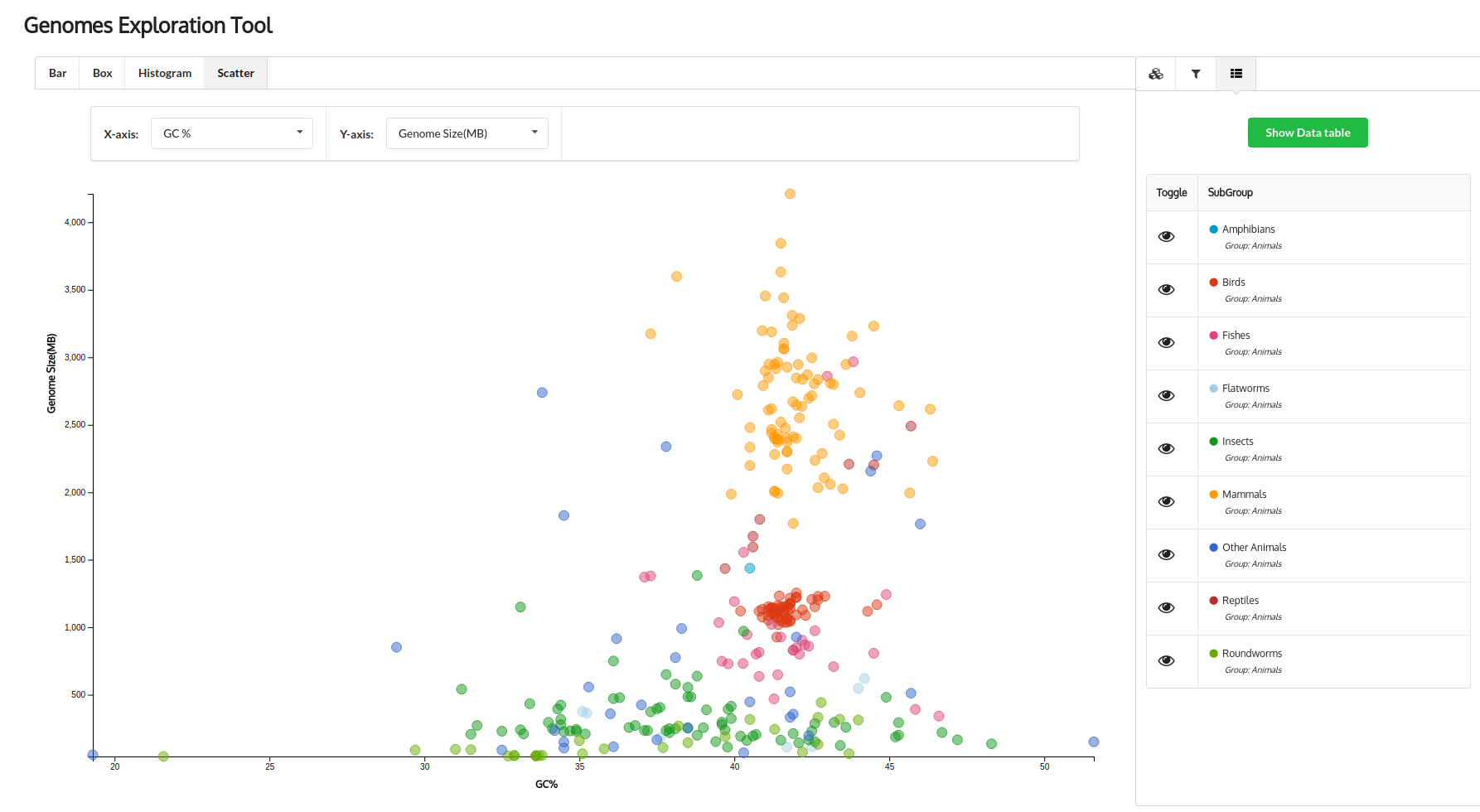
The
NCBI Genome database is a collection of information on all the genome sequencing projects done and are
in progress till date. NCBI provides a very basic browsing interface, which represents the whole data in a
tabular format and has limited options for a user to explore through the genomes and find the information they
need. To provide a user-friendly and interactive tool to explore this data, we developed Genomes Exploration
Tool (GET) using a JavaScript plotting library called d3.js. GET uses data provided by NCBI and converts it
into clear, interactive and visually appealing plots, which can be navigated and interacted with as the user
desires. Using attributes such as Genome size, GC content, Number of genes and proteins etc. for more than
18000 genomes, users can explore the genomes using bar plots, scatter plots, box-and-whisker plots and histograms.
MSDB -
A comprehensive database of microsatellites
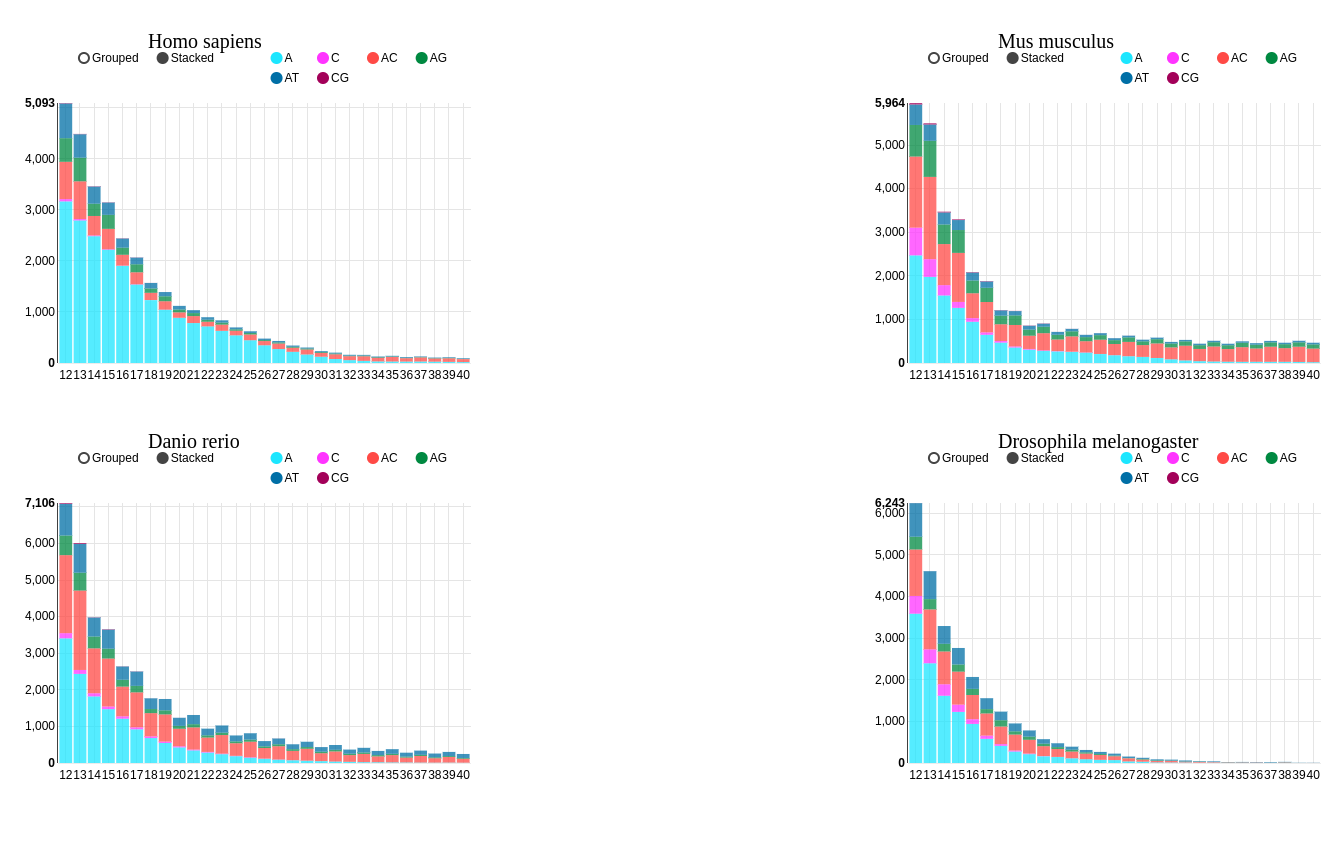
Microsatellites, also known as Simple Sequence Repeats (SSRs), are short tandem repeats of 1–6 nt motifs present in all genomes,
particularly eukaryotes. Besides their usefulness as genome markers, SSRs have been shown to perform important
regulatory functions, and variations in their length at coding regions are linked to several disorders in humans.
Microsatellites show a taxon-specific enrichment in eukaryotic genomes, and some may be functional. MSDB (Microsatellite
Database) is a collection of >650 million SSRs from 6,893 species including Bacteria, Archaea, Fungi, Plants,
and Animals. This database is by far the most exhaustive resource to access and analyze SSR data of multiple
species. In addition to exploring data in a customizable tabular format, users can view and compare the data
of multiple species simultaneously using our interactive plotting system. MSDB is developed using the Django
framework and MySQL.
PERF -
Perfect, Exhaustive Repeat Finder
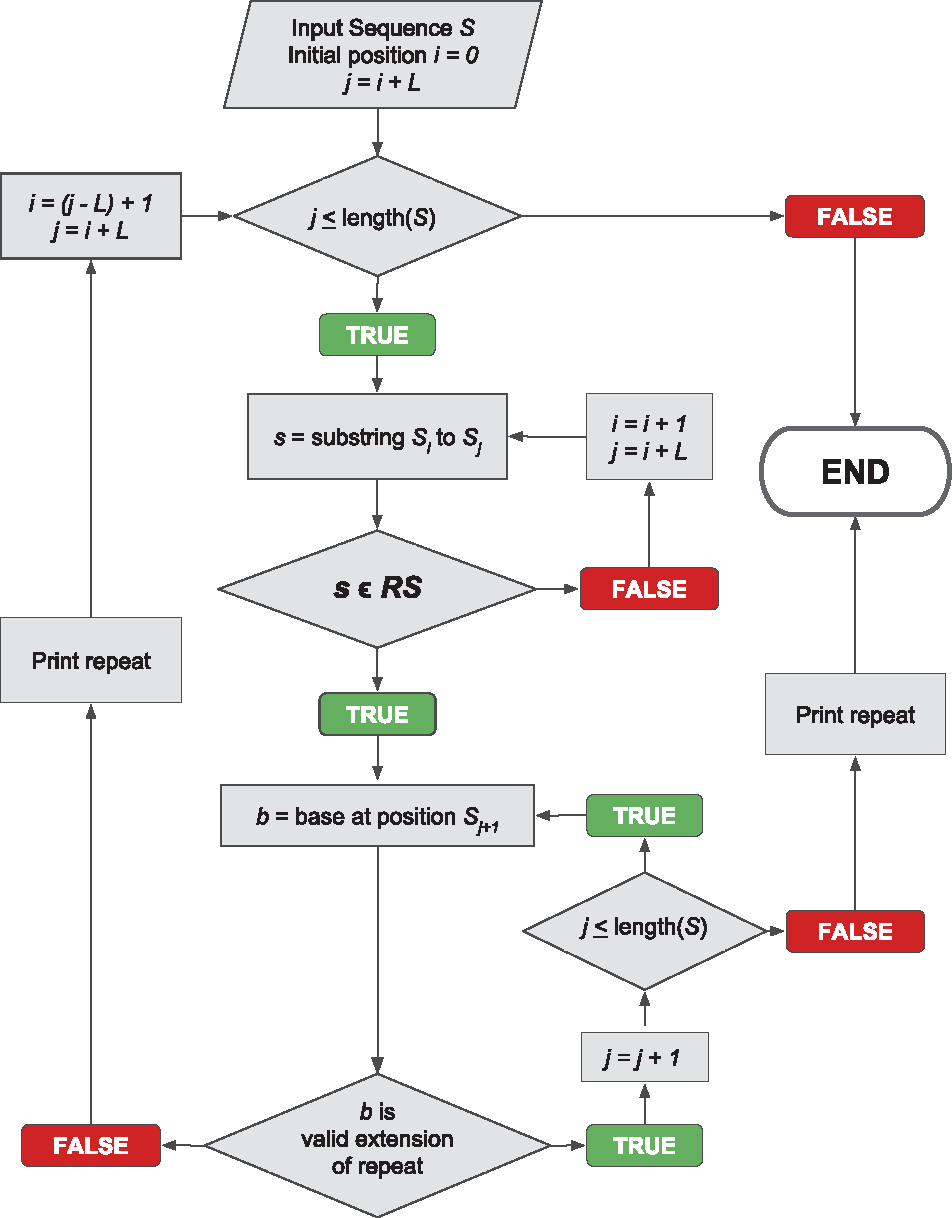
PERF is a Python package developed for fast and accurate identification of microsatellites (SSRs) from DNA sequences. The existing tools for SSR identification have one or more caveats in terms of speed, comprehensiveness, accuracy, ease-of-use, flexibility and memory usage. PERF was designed to address all these problems.
PERF is a recursive acronym that stands for "PERF is an Exhaustive Repeat Finder". It is compatible with both Python 2 (tested on Python 2.7) and 3 (tested on Python 3.5). Its key features are:
- Fast run time, despite being a single-threaded application. As an example, identification of all SSRs from the entire human genome takes less than 7 minutes. The speed can be further improved ~3 to 4 fold using PyPy (human genome finishes in less than 2 minutes using PyPy v5.8.0)
- Linear time and space complexity (O(n))
- Identifies perfect SSRs
- 100% accurate and comprehensive - Does not miss any repeats or does not pick any incorrect ones
- Easy to use - The only required argument is the input DNA sequence in FASTA format
- Flexible - Most of the parameters are customizable by the user at runtime
- Repeat cutoffs can be specified either in terms of the total repeat length or in terms of number of repeating units
- TSV output and HTML report. The default output is an easily parseable and exportable tab-separated format. Optionally, PERF also generates an interactive HTML report that depicts trends in repeat data as concise charts and tables