Molecular Chaperones
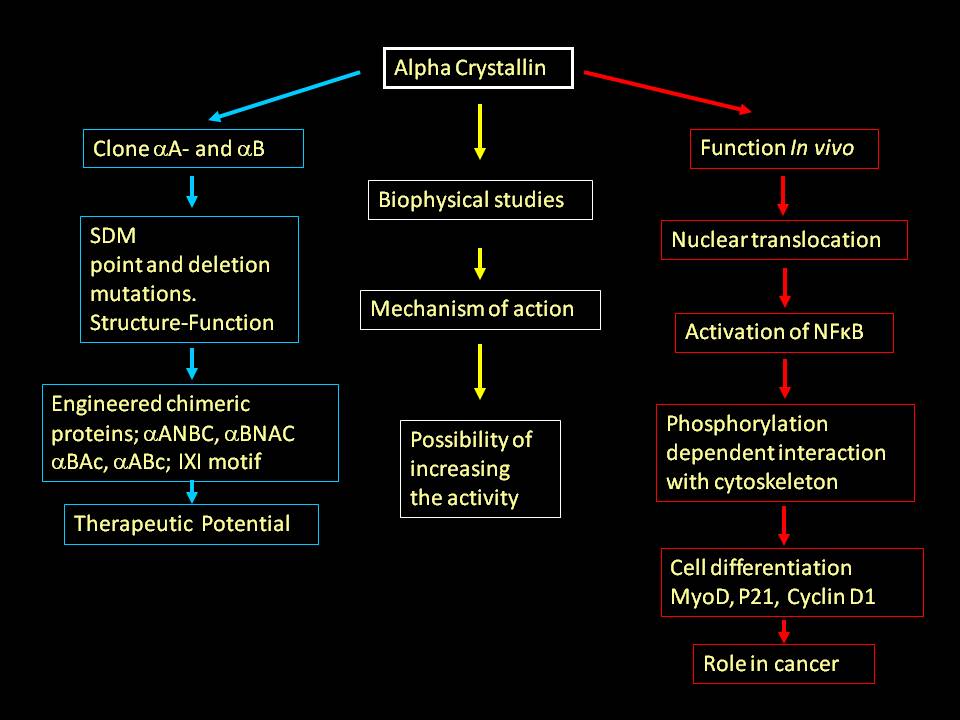

We have shown that temperature dependent structural perturbation leads to enhanced activity of a small heat shock protein, α-crystallin (J.Biol.Chem. 1994). Our studies suggested that the loss of chaperone activity is the molecular basis for desmin related myopathy associated with R120G and cataracts associated with R116C mutations in &alpha crystallin (J. Biol. Chem., 1999). Our biophysical and molecular biological investigations of α-crystallin have shown structure-function relationship and possibility of increasing the activity by structural alterations (series publications in J. Biol. Chem. and a few others FEBS Letters, Biochem. J etc). This finding opened up the possibility of mitigating the complications arising out of protein misfolding and aggregation by enhancing chaperone-like activity.
Utilizing molecular biology tools we have engineered chimeric proteins with several fold higher activity with potential therapeutic applications (J. Biol. Chem., 2000, 2002, 2003) (we have patented one of these engineered proteins). We showed that a biocompatible, small molecule can enhance the chaperone activity. Interestingly, this molecule can also help mutant protein (R120G, that leads to desmin related myopathy) to gain almost about 80% of the activity. Our later, cell biological, studies showed the possibility of α-crystallin’s role in stabilizing the nuclear matrix (Exp. Cell Res., 2004), transcriptional apparatus and cytoskeletal organization (J. Mol. Biol., 2007).
Recently a cataract causing mutation (G98R) is reported from India. Our study showed that the mutation leads to folding defective, aggregation prone protein. Despite such deleterious protein the cataract is pre-senile and not congenital; our studies have shown that the mixed oligomer formation to be responsible for the absence of congenital cataract (Mol. Vision, 2006. J. Mol. Biol., 2007). The small heat shock proteins are critical in several cellular processes. These proteins are known to work in concert. Human genome has 10 small heat shock genes. We are working on HspB2, HspB3, HspB4, HspB5 and HspB8, and have initiated work on HspB1 and HspB6 as well. We have shown that HspB8, earlier known as hsp22, has a putative heat shock factor-1 binding region (upstream of coding region) and demonstrated that indeed HspB8 can be over expressed at heat shock temperatures (Biochem. J., 2004, 2007).
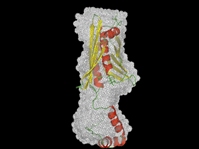
HSP33 is an interesting heat shock protein under redox control. Hsp33 in vivo exists as a reduced molecule with no activity. Upon oxidative stress, it becomes active and prevents aggregation of proteins. We have shown that oxidative activation of hsp33 involves conformational change accompanying significant increase in the surface hydrophobicity. This provided the molecular basis for the oxidative activation. The two reported crystal structures of Hsp33 lack about 60 amino acids at the C-terminus. We have carried out small angle X-ray scattering studies of the full-length protein and determined the abinitio shape profile. Based on these, we could suggest that the C terminal region can fold over and prevent inter-protein interaction. Oxidation leads to opening of this region and form active dimers (J. Biol. Chem., 2004)
