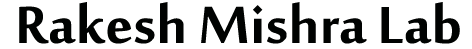The cells of a multi-cellular organism contains the same genetic blueprint. However, it is the selective expression of specific sets of genes that leads to specific cell types. The packaging of the genome in a specific manner allows silenced genes to be hidden from the transcriptional machinery while making the expressed genes more accessible to the same. What is the “genome packaging code” that allows packaging of the genome into cell type specific forms? We are interested in studying the genome organization responsible for this differential and regulated expression of genetic information.
I. Genome organization and gene regulation
Chromatinization of the genome not only allows it to fit in the nucleus, but also ensures that only a subset of genes are accessible to the transcription apparatus. We are interested in the DNA elements that maintain the boundary between different states of chromatin (hence the expression status), as well as the elements that not only maintain the expression state of a cell but also transmit it after the cell divisions.
Chromatin boundaries subdivide a genome into different expression states, either by blocking enhancers/ silencers, or by preventing the spread of heterochromatin. Not enough is known about these elements to identify them in the genome de novo to understand their modus operandi. Our lab have contributed in deciphering the mechanism of the functioning of such cis regulatory elements.
Boundary elements demarcate functionally independent domains of closely spaced but differentially expressed genes.
We identified a chromatin boundary element, the ME boundary between adjacent genes - myoglianin and eyeless - that are differentially expressed in Drosophila. ME boundary requires BEAF (Boundary Element Associated Factor) and GAGA Associated Factor to maintain the functionally autonomous chromatin domains(NAR, 2011).
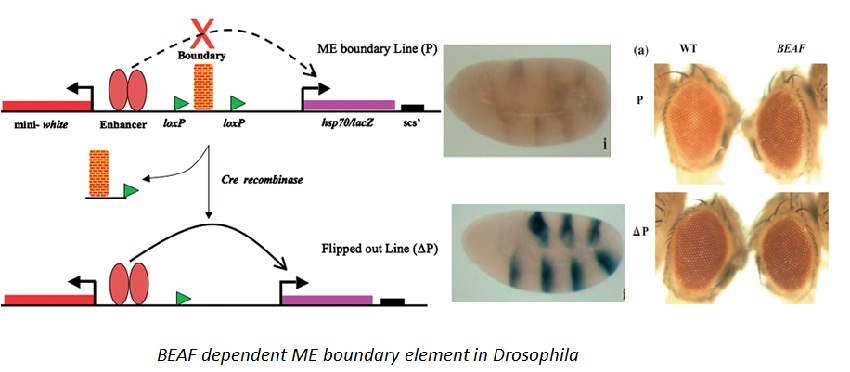
Genome-wide prediction of boundary elements in insects: We developed a tool to predict boundary elements in Drosophila. Our comparative analysis also revealed that like Drosophila, the Bithorax region in insects like mosquito (A. gambiae ) contains an extensive array of boundaries that act as enhancer blocker in Drosophila indicating that they are evolutionarily conserved. [NAR, 2013]
Cellular memory systems: One of the major unanswered question in the field of genome biology is how the cell remembers the expression state and the corresponding organization of genome each time it divides. Regulatory elements referred to as Cellular memory elements, Polycomb response elements (PREs) / Trithorax response elements (TREs) are responsible for maintaining the expression state of a genome. Several such elements have been identified in Drosophila. Failure of these systems can lead to mis-expression resulting in developmental defects and diseases. We have shown that Trl-GAGA plays a role in maintaining the repressed state of target genes involving lolal, which may function as a mediator to recruit PcG complexes.
II. Expansion of epigeneti tool kit during vertebrate evolution
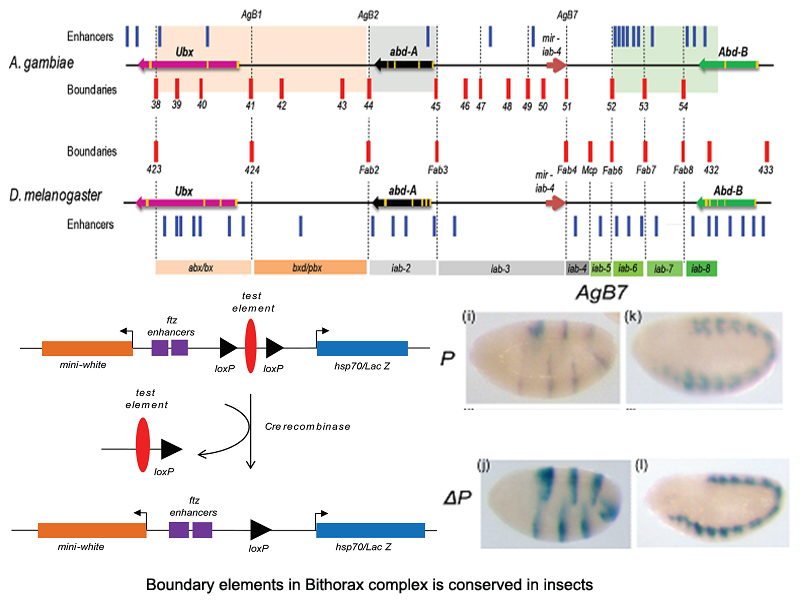
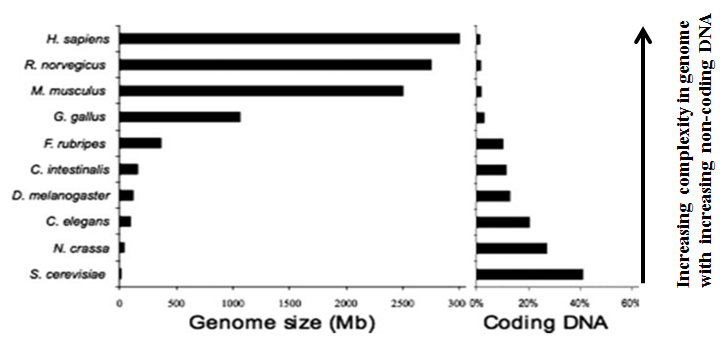
While the human genome is 20 fold larger than that of Drosophila, it contains only 1.5 times more genes. As in the case with the cis elements like PREs, it is also expected that the trans acting factors PcG/trxG binding to PRE/TREs must also be conserved from flies to mammals. This indicates that the PcG epigenetic toolkit has expanded in the vertebrates. Each fly gene has 2-7 vertebrate homologues. Most of these novel genes encode transcription factors involved in gene regulation. Thus, the expansion in the genome size is due to the addition of new regulatory features in the existing genes rather than addition of new genes.
Using comparative analysis of protein and genome sequence database of various organisms for around 100 PC homologues we have identified a novel DNA binding motif - AT-Hook adjacent to the chromodomain, in all the vertebrate homologues. This AT-Hook can restrict nucleosome dynamics via a three-way 'PC-histoneH3-DNA' interaction. Our findings represent an expansion of epigenetic toolkit along side the evolution of complexity. [BMC Genomics 2009]
III. Functional relevance of non-coding DNA
A large proportion of the genome in higher eukaryotes consists of non-coding DNA. Since such DNA accumulated and persisted during evolution, implies that there should be a functional relevance for the non-protein coding DNA, which is increasingly being appreciated in the genomic era. We address the functional relevance of non-coding genome by bioinformatics, molecular biology and genetic approaches.
We are interested in different kinds of non-coding DNA sequences for their functional relevance.
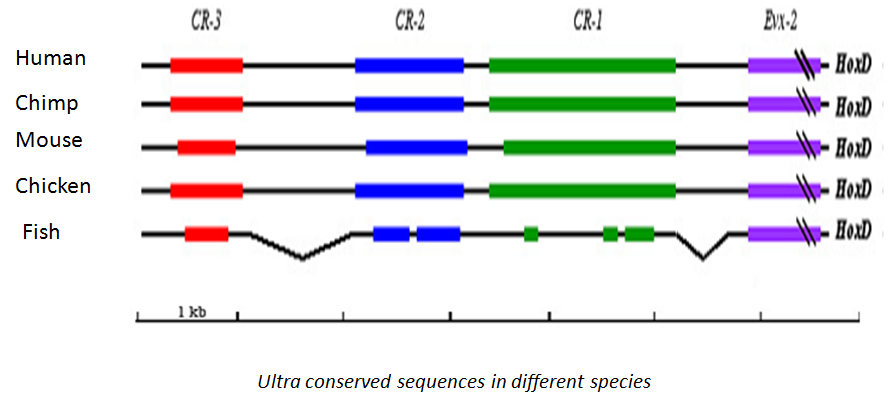
CNCS (conserved non coding sequences)
By comparing genomes of different species, regions that have been conserved can be identified. Such conservation can be indicative of a functional relevance. Comparisons among vertebrates have led to the identification of long regions that show nearly 100% identity. In humans, such "ultra-conserved regions" make up ~3% of the genome, more than even the coding fraction. Such remarkable conservation indicates that these regions are under tremendous selection pressure. In most cases, CNCS are present near developmentally regulated genes. However, their functions are yet to be well established.
We are interested in understanding the functional significance of "ultra conserved region" – CR1,2,3 upstream HoxD locus which was the first reported in vertebrates by our lab. [Genome Biology 2003]
SSR (simple sequence repeats)
SSRs are repeats of 1-6 base-pair units of DNA. Not all SSRs possess a random distribution of their location and some are highly enriched in complex genomes. Some show a higher abundance of longer repeats as opposed to shorter repeats, which is unusual.
Using bioinformatics approach, we analysed all possible SSRs combinations exhaustively across 24 eukayotic genomes, and reported that only 73 SSRs of 501 possible combinations show length specific enrichment.
One of the interesting finding of such studies was that only in few SSRs larger size repeats are more abundant than the shorter ones. This accumulation of large sized repeats of a preferred size repeat must have some functional significance. [Gene, 2014].
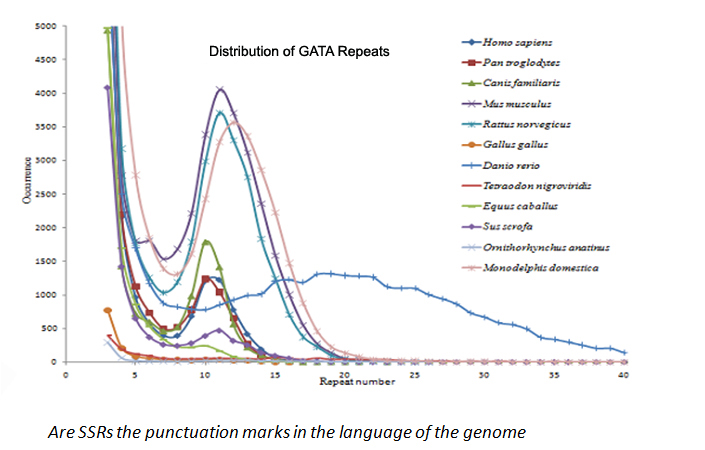
One such SSR repeat where the longer size repeat is more abundant than shorter one is the tetra-nucleotide repeat GATA. (GATA)10-12 is far more abundant than (GATA)6. This implies that longer repeats of it have been enriched and have provided an advantage to the organism. Our recent findings suggest GATA acts an enhancer blocker in flies and human cells. [Nature Communications, 2013]

Patterns and motif clusters
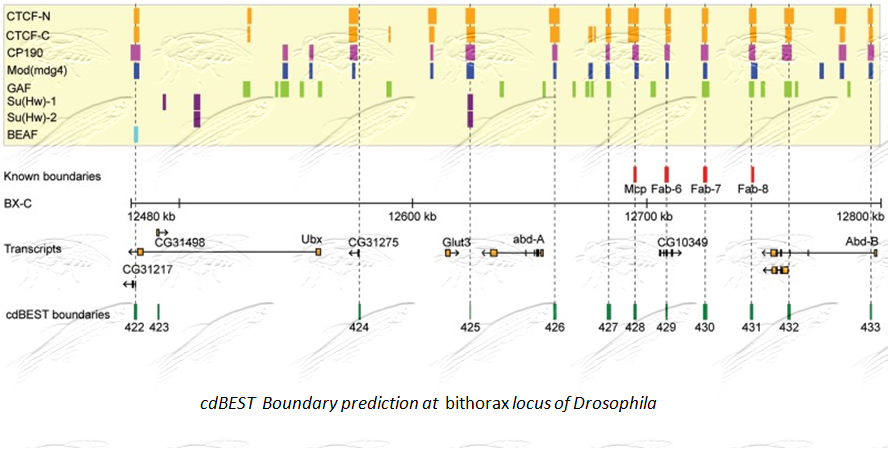
About one third of the genome is unique as it can not be grouped in any common class. Using the power of novel bioinformatics and high throughput techniques we seek to unravel the various patterns and motif clusters that designate the various cis regulatory elements particularly Boundary Elements.
We have developed a boundary element predicting tool cdBEST (chromatin domain Boundary Element Search Tool) that identifies boundary elements in Drosophila species. [NAR, 2012]
|